Reduktion af kompleksitet i biologiske data: Anvendelse af PCA i mikrobiomestudier#
Af Adam Greve Bregenhof og Christian Adam Deding Nielsen. Under vejledning af Jakob Lemvig og Mikael Lenz Strube.
Introduktion#
Inden for bioengineering og bioteknologi arbejder man ofte med målinger, der producerer store mængder data. Det kan være genekspressionspaneler, proteomik, metabolomik, mikroskopikarakteristika eller mikrobielle samfundsprofiler (f.eks. 16S-/amplicon-sekventering). Disse datasæt indeholder let hundreder til tusinder af variabler pr. prøve, mens antallet af prøver ofte er langt mindre.
En tilbagevendende opgave er at finde skjulte strukturer eller beskrivende mønstre i data. Dette kan for eksempel være:
at identificere latente grupperinger (f.eks. patientundertyper, behandlingsresponsgrupper eller økologiske nicher),
at undersøge, om prøvedata klumper sig sammen efter miljø/geografi (f.eks. jordprøver fra forskellige lokaliteter),
og at identificere hvilke variabler (gener, taxa, karakteristika), der driver adskillelse og gruppering i data, for at øge forklarbarheden.
Her bliver dimensionsreduktion nyttig. Den komprimerer data til få informative koordinater, som vi kan visualisere og fortolke. I dette projekt vil du undersøge de matematiske idéer bag to almindelige metoder:
PCA (Principal Component Analysis / hovedkomponentanalyse): finder retninger med maksimal varians baseret på kovarians-/korrelationsstrukturen i data.
PCoA (Principal Coordinate Analysis / klassisk MDS): finder lavdimensionelle koordinater ud fra en valgt afstand mellem prøver (nyttigt når en domænespecifik forskel er mere meningsfuld end de rå karakteristika).
Til sidst vil du anvende disse metoder på et virkeligt eksempel med mikrobiomer. Mikrobiomet er samlingen af mikroorganismer, der lever i et habitat, såsom menneskets tarm eller et jordmiljø. Mikrobiomets sammensætning er knyttet til sundhed og sygdom, og store studier bruger ofte mikrobiomprofiler som et “fingeraftryk”, der kan adskille grupper (f.eks. raske vs. sygdomsassocierede samfund). Lignende principper gælder for jord: forskellige lokaliteter og forhold (vegetation, næringsstoffer, fugtighed, menneskelig påvirkning) kan føre til markant forskellige mikrobielle samfund. Dimensionsreduktion gør det muligt at se sådanne mønstre ved at komprimere hundreder af taxa/karakteristika til få fortolkelige akser (nogle gange kan en akse fortolkes som “høj vs. lav forekomst af bestemt taxon”).
I afsnit 1–2 opbygger vi intuition og implementerer PCA fra bunden på mindre datasæt, og i afsnit 3–4 udvider vi idéerne til PCoA og til virkelige mikrobiomdata.
Projektmål#
Du skal skrive en sammenhængende rapport om PCA og PCoA samt deres anvendelse på biologiske datasæt. Du kan bruge spørgsmålene i denne notebook som vejledning, men du behøver ikke at henvise til dem direkte. Spørgsmål markeret med * er mere udfordrende. I nogle spørgsmål bliver du bedt om at bevise noget, og i disse tilfælde kan det være en fordel at konstruere specifikke eksempler – f.eks. hvis du bliver bedt om at vise, at en matrix \(A\) har positive egenværdier, kan du konstruere matricer i SymPy/NumPy med samme struktur som \(A\) og beregne egenværdierne. Vi opfordrer dig naturligvis også til i det mindste at forsøge at lave et formelt bevis. I rapporten kan du vælge at fokusere mere på den biologiske del ved at inddrage forskellige datasæt og diskutere anvendeligheden af PCA/PCoA i denne sammenhæng (afsnit 1 og 4). Du kan også vælge at fokusere mere på den matematiske teori, f.eks. ved at arbejde med PCA for komplekse data og/eller ved at vise, at PCA og PCoA i visse tilfælde er ækvivalente (afsnit 2 og 3).
Motivationstjek: mikrobiomer og miljøprøver#
Opgave 0.1#
Forklar kort, med dine egne ord, hvad et mikrobiom er, og hvad man typisk måler, når man “profilerer” et mikrobiom?
Opgave 0.2#
Jordprøver fra forskellige lokaliteter kan have meget forskellige mikrobiomer. Angiv to plausible biologiske/miljømæssige grunde til dette.
Opgave 0.3#
Antag, at du tager jordprøver i Dyrehaven samt et andet sted i Danmark og får hundreder af taxa pr. prøve. Forklar (konceptuelt), hvordan PCA/PCoA kan hjælpe dig med at se, om lokaliteterne er forskellige.
Forberedelse#
Egenværdier, egenvektorer og diagonalisering: afsnit 12.1-12.3 (Matematik 1a)
Diagonalisérbare matricer, spektralsætningen, projektioner: afsnit 2.7-2.10 (Matematik 1b)
Tema 2: Data-matricer og dimensionsreduktion
Projektmål#
Når du har gennemført dette projekt, bør du være i stand til at:
forklare, hvorfor højdimensionelle biologiske data ofte har gavn af dimensionsreduktion.
standardisere og centrere data, danne kovarians- (eller korrelations-)matricer og beregne hovedkomponenter.
fortolke egenværdier og egenvektorer i relation til varians og bidrag fra biologiske karakteristika.
forklare forskellen mellem karakteristik-baserede metoder (PCA) og afstandsbaserede metoder (PCoA), samt hvornår hver metode er passende.
anvende PCA/PCoA på biologiske datasæt (Iris-blomster og mikrobiom-/jordprøver) og diskutere de observerede klyngedannelser.
Afsnit 1: Opvarmning: Datamatricen og PCA i NumPy#
Biologisk datasæt til opvarmning: Iris-blomster#
Før vi dykker ned i den generelle matrixnotation, starter vi med et klassisk lille biologisk datasæt: Iris-datasættet (R. A. Fisher, 1936). Hver prøve er en blomst, beskrevet ved fire målte karakteristika:
bægerbladslængde (eng: sepal length) i cm
bægerbladsbredde (eng: sepal width) i cm
kronbladslængde (eng: petal length) i cm
kronbladsbredde (eng: petal width) i cm
samt en artsbetegnelse (setosa, versicolor, virginica).
Selv her, med kun 4 karakteristika, er dimensionsreduktion nyttig: det gør det muligt at visualisere data i 2D/3D og se, om arterne danner adskilte klynger, samt hvilke karakteristika, der driver adskillelsen.
Du kan læse mere om Iris-datasættet her: Iris Data
Opgave 1.0#
(a) Hvad repræsenterer variablerne (karakteristikaerne) biologisk? (b) Hvorfor kan det være nyttigt at reducere data fra 4 dimensioner til 2 dimensioner, før man opbygger en klassifikationsmodel?
Hvis du ikke har scikit-learn installeret, kan det gøres i terminalen med conda install scikit-learn.
import numpy as np
import matplotlib.pyplot as plt
from sklearn.datasets import load_iris
iris = load_iris()
X_iris = iris['data'] # (150, 4)
y_iris = iris['target'] # (150,)
feature_names = iris['feature_names']
target_names = iris['target_names']
print('X_iris shape:', X_iris.shape)
print('Features:', feature_names)
print('Classes:', target_names)
X_iris shape: (150, 4)
Features: ['sepal length (cm)', 'sepal width (cm)', 'petal length (cm)', 'petal width (cm)']
Classes: ['setosa' 'versicolor' 'virginica']
Før vi begynder vores udforskning af Iris-datasættet, vil vi genopfriske nogle centrale emner fra Tema 2: Data-matricer og Dimensionsreduktion, herunder datamatricen, kovariansmatricen samt essentielle NumPy-funktionaliteter til at udføre PCA på et lille datasæt. Det kan være nyttigt at vende tilbage til Ekstra Frivillig Opgave: PCA på Iris-datasættet (4D) inden du går videre.
Vi betragter en vilkårlig datamatrix \(X \in \mathbb{R}^{n \times d}\), hvor \(n\) angiver antallet af datapunkter, og \(d\) repræsenterer antallet af karakteristika, eller ækvivalent dimensionaliteten. Vi ønsker at standardisere vores data ved at fratrække middelværdien \(\mu_1, \mu_2, \dots, \mu_d\) for hver kolonne, hvilket kan skrives som
hvor \(\pmb{\mu} \in \mathbb{R}^d\) og \(\pmb{c} \in \mathbb{R}^n\).
Opgave 1.1#
Opskriv definitionen på \(\pmb{\mu}\) og \(\pmb{c}\), således at \(\hat{X}\) bliver til den standardiserede matrix.
Fra \(\hat{X}\) skal vi beregne kovariansmatricen
Opgave 1.2a#
Angiv størrelsen af matricen \(C\), og vis, at den tillader en spektral dekomposition på formen
\[\begin{equation*} C = Q \Lambda Q^T, \end{equation*}\]hvor \(Q\) består af \(C\)’s egenvektorer og \(\Lambda\) af dens egenværdier.
Opgave 1.2b*#
Vis, at \(C\) og \(\Lambda\) begge er positiv semidefinit (PSD) og dermed ikke har nogen negative egenværdier. Hvis \(\hat{X}\) har fuld rang, har \(C\) så nogen egenværdier lig nul?
Hint: I sidste spørgsmål må I gerne bruge uden bevis at rangen af en vilkårlig matrix \(A\) er lig med rangen af \(A A^T\) (og \(A^T A\)).
Selvom det er mindre almindeligt, er der intet til hinder for også at betragte det tilfælde, hvor data er komplekse, dvs. at \(X \in \mathbb{C}^{n \times d}\). I dette tilfælde ønsker vi en hermitisk kovariansmatrix.
Opgave 1.3#
Husk på, hvad det vil sige for en matrix at være hermitisk, og opskriv definitionen på kovariansmatricen \(C\) for en kompleks datamatrix \(X\).
\[\begin{equation*} C = \text{??} \end{equation*}\]Hvis \(X\) er kompleks, kan \(C\) også være kompleks. Gælder det stadig at egenværdierne for kovariansmatricen \(C\) er ikke-negative, reelle tal?
Visualisering af data#
På baggrund af den spektrale dekomposition af \(C\) kan vi konkludere, at den har reelle egenværdier og ortogonale egenvektorer, hvilket er nyttigt til projektion af data. Før vi kaster os ud i projicering, vil vi se på, hvordan vi kan implementere vores datateknikker i NumPy. Vi starter med kun at plotte to dimensioner af Iris-dataene, uden PCA, og farvelægger efter art.
import numpy as np
import matplotlib.pyplot as plt
from sklearn.datasets import load_iris
iris = load_iris()
X_full = iris["data"]
y = iris["target"]
feature_names = iris["feature_names"]
target_names = iris["target_names"]
feat_idx = [0, 1]
X = X_full[:, feat_idx]
plt.figure(figsize=(6, 5))
for c in np.unique(y):
plt.scatter(
X[y == c, 0], X[y == c, 1],
s=70, edgecolors="white", linewidth=1.0, alpha=0.85,
label=target_names[c]
)
plt.title("Iris: 2D plot farvet efter art", fontsize=14, weight="bold")
plt.xlabel(feature_names[feat_idx[0]])
plt.ylabel(feature_names[feat_idx[1]])
plt.grid(True, linestyle="--", alpha=0.5)
plt.legend()
plt.tight_layout()
plt.show()
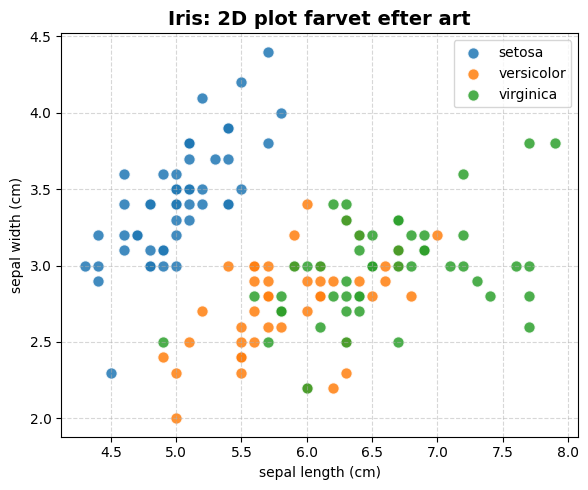
Opgave 1.4#
Plottet ovenfor vil ikke give særlig meget mening (bortset fra at vi muligvis kan skelne setosa-arten fra de andre). Lad os prøve at tilføje endnu en dimension til datapunkterne og lave et tilsvarende plot i 3D.
Nu begynder vi at se lidt mere klyngedannelse. Men da vi ikke kan visualisere flere dimensioner, går informationen fra den sidste dimension i data tabt i vores analyse. Det er her PCA kommer ind i billedet og gør det muligt at reducere data til et antal dimensioner, der kan visualiseres.
Visualisering af kompleks PCA *#
Hvis det bliver relevant, kan visualisering af komplekse data udføres på flere måder:
Punktplot (scatter plot) af realdel vs. imaginærdel
Plot af størrelse (magnitude) vs. fase
Disse repræsentationer hjælper med os med at fortolke datastrukturen i det komplekse rum.
"""
Visualisér kompleks PCA-projektion som real- vs imaginærdel med punktplot.
"""
import matplotlib.pyplot as plt
def plot_complex_pca_projection(X_proj, title="Kompleks PCA-projektion"):
plt.figure(figsize=(6, 5))
plt.scatter(
X_proj.real,
X_proj.imag,
s=80,
edgecolors="white",
linewidth=1.2,
alpha=0.85
)
plt.title(title, fontsize=14, weight="bold")
plt.xlabel("Realdel af PC1", fontsize=12)
plt.ylabel("Imaginærdel af PC1", fontsize=12)
plt.grid(True, linestyle="--", alpha=0.5)
plt.axis("equal")
# Pæn kant-styling
for spine in ["top", "right"]:
plt.gca().spines[spine].set_visible(False)
plt.tight_layout()
plt.show()
"""
Visualisér kompleks PCA-komponent i polær form (fase vs størrelse).
"""
import numpy as np
import matplotlib.pyplot as plt
def plot_complex_polar(X_proj, pc=0):
# Beregn størrelse og fase af de valgte hovedkomponenter
magnitude = np.abs(X_proj)
phase = np.angle(X_proj)
plt.figure(figsize=(6, 5))
plt.scatter(
phase,
magnitude,
s=80,
edgecolors="white",
linewidth=1.2,
alpha=0.85
)
plt.title(f"Fase-størrelse af PC{pc+1}", fontsize=14, weight="bold")
plt.xlabel("Fase (radianer)", fontsize=12)
plt.ylabel("Størrelse", fontsize=12)
plt.grid(True, linestyle="--", alpha=0.5)
# Pæn kant-styling
for spine in ["top", "right"]:
plt.gca().spines[spine].set_visible(False)
plt.tight_layout()
plt.show()
Opgave 1.5#
Brug følgende funktionsskabeloner til at udføre den spektrale dekomposition ud fra datamatricen \(X\) (ikke-standardiseret). (Hint: Egenværdidekompositionen for en hermitisk matrix håndteres lidt anderledes i NumPy. Se dokumentationen for
numpy.linalg.)
def center_columns(X):
"""Centrér hver søjle i X
Input: X : (n_samples x n_features)-matrix
Output: Xc: (n_samples x n_features)-matrix med nulmiddel-søjler
"""
# TODO (essentiel): Beregn søjlemiddelværdier og træk fra.
raise NotImplementedError("Beregn søjlemiddelværdier og træk fra X.")
def covariance_matrix(Xc):
"""Dan kovariansmatrix med centreret data Xc.
Input: Xc: (n x d) centreret
Output: CC: (d x d)-kovariansmatrix
"""
n = Xc.shape[0]
# TODO (essentiel): Implementér den sædvanlige uvildige (unbiased) kovariansmatrix.
raise NotImplementedError("Returnér prøve-kovariansmatricen.")
def eig_sorted_symmetric(C):
"""Egendekomposition af en symmetrisk matrix C.
Input: C: Kovariansmatrix
Returnerer (vals, vecs), hvor vals er sorteret aftagende, og søjlerne i vecs er ordnet tilsvarende.
TODO (essentiel): Lav egendekompositionen og sortér.
Hints:
- eigenvects() returnerer [(egenværdi, multiplicitet, [basisvektorer...]), ...]
- Opbyg 'vals' som en liste og 'vecs' som en matrix med egenvektorer som søljer.
"""
# TODO: Beregn og sortér egenpar
raise NotImplementedError("Beregn sorterede egenpar for symmetriske matricer.")
Test din implementation ved at plotte de tilsvarende egenrum for vores lille testeksempel:
Visualisering af data#
Vi er nu klar til at udføre projektionen. Ideen bag PCA er, at de største af \(C\)’s egenværdier svarer til egenvektorer med retninger, der indeholder mest mulig information eller varians i vores data. For et givet \(k < d\) vil vi konstruere projektionsmatricen \(P\), således at \(P\pmb{x}\) projicerer et datapunkt \(\pmb{x}\) ned på underrummet udspændt af de \(k\) egenvektorer \(\pmb{e_1}, \dots, \pmb{e_k}\) med de største egenværdier. Husk definitionen af en sådan projektion:
Opgave 1.6#
Opstil ud fra definitionen ovenfor et udtryk for projektionsmatricen \(P\), og angiv, hvordan alle datapunkter kan projiceres med én enkelt matrixmultiplikation. Husk, at vi også betragter komplekse data.
Opgave 1.7#
Udfyld følgende funktionsskabelon, så den implementerer hele PCA-arbejdsgangen: Givet en vilkårlig datamatrix og underrumsdimension \(k\) skal outputtet være de projicerede datapunkter samlet i en matrix \(Z\) samt de \(k\) egenværdier \(\lambda_1, \dots, \lambda_k\). Plot de projicerede datapunkter i samme figur som i opgave 3. Hvad kan du sige om de klynger, du ser, og kan du finde en forklaring i data på disse klynger, dvs. er der en karakteristik, der bestemmer disse klynger?
def pca_from_cov(X, k: int):
"""Hovedkomponentanalyse bed brug af kovarians egendekomposition.
Trin:
1) Centrér X -> Xc
2) Beregn kovariansmatrix C
3) Egendekomponér C -> egenværdier (lambdaer) & egenvektorer (retninger)
4) Sortér egenvektorer for faldende varians
5) Behold top-k komponenter og opbyg projektionsmatrix -> P
6) Projicér ned i det k-dimensionelle underrum -> Z-matrix
Returnerer: Z, lambdaer
TODO (essentiel): Kald din hjælpefunktion og saml outputs.
"""
raise NotImplementedError("Saml PCA fra kovarians egendekomposition.")
Afsnit 2: PCA som et optimiseringsproblem#
I denne opgave vil vi arbejde med PCA fra et optimeringsperspektiv og koble det til en egenværdiformulering. Du ved allerede, at PCA kan beregnes ved hjælp af en spektral dekomposition af kovariansmatricen. Men hvad gør PCA egentlig? I dette afsnit vil vi få at se, at PCA også kan betragtes som et optimeringsproblem, hvor man finder den hovedretning, der indfanger så meget varians i dataene som muligt.
Centrering og projicering af data#
Kør cellen herunder for at generere nogle prøver af 2D data og centrere det.
import numpy as np
import matplotlib.pyplot as plt
# Dan 2D-prøvedata
np.random.seed(0)
X_big = np.random.randn(100, 2) @ np.array([[2, 1], [1, 1]]) # Korrelerede data
# Centrér dataene (fratræk søjlemiddelværdier)
X_big = X_big - X_big.mean(axis=0)
plt.scatter(X_big[:,0], X_big[:,1], alpha=0.7, edgecolors='white', linewidth=1)
plt.axis('equal')
plt.title("Centrerede datapunkter")
plt.xlabel("$x_1$")
plt.ylabel("$x_2$")
plt.grid(True, linestyle='--', alpha=0.5)
plt.show()
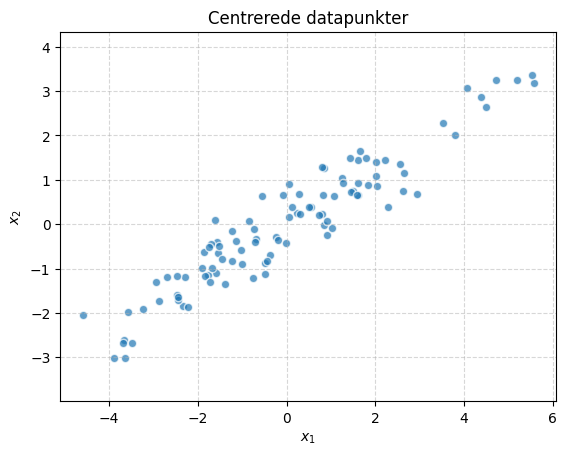
Hver række i \(X\) er et datapunkt \(\pmb{x}_i\). Projicerer vi hvert punkt ind på en retningsvektor \(\pmb{w}\), er projektionen givet ved:
Opgave 2.1#
Opskriv et NumPy-udtryk herunder, der beregner alle projektioner på én gang (vektoriseret).
# Beregn alle projektioner z = X @ w
w = np.array([1, 0]) # Prøv først med x1-retningen
# z = ...
Variansmåling langs en retning#
Variansen af the projicerede data langs en retning \(( \pmb{w} )\) er:
Lad os beregne den for et par tilfældige retninger:
def variance_in_direction(X, w):
w = w / np.linalg.norm(w)
return (np.linalg.norm(X @ w)**2) / X.shape[0]
for angle in [0, 30, 60, 90]:
theta = np.deg2rad(angle)
w = np.array([np.cos(theta), np.sin(theta)])
print(f"Vinkel {angle:>2}° → Varians = {variance_in_direction(X_big, w):.3f}")
Vinkel 0° → Varians = 5.110
Vinkel 30° → Varians = 6.977
Vinkel 60° → Varians = 5.435
Vinkel 90° → Varians = 2.026
Opgave 2.2#
Prøv at generere flere tilfældige \(w\)-vektorer, normalisér dem, og beregn variansen hver gang. Hvilken retning (vinkel) ser ud til at give den største varians? Prøv også at plotte vektorerne sammen med datapunkterne.
For at vores valgte projektion af datapunkter skal beskrive dataene bedst muligt, bør vi derfor forsøge at maksimere variansen af de projicerede punkter på retningsvektoren (dvs. sprede punkterne så meget som muligt langs projektionen). I PCA kaldes denne retningsvektor, som maksimerer variansen, en hovedkomponent (Principal Component), og vi kan nu formulere målet med at finde disse hovedkomponenter formelt som:
Opgave 2.3a#
Hvorfor er denne betingelse \(( \| \pmb{w} \| = 1 )\) nødvendig? Hvad ville der ske, hvis vi så bort fra den?
SVD og hvorfor den optræder i PCA#
Singulærværdi-dekompositionen (SVD) faktoriserer enhver matrix
\(X \in \mathbb{R}^{n \times d}\) som
hvor \(U \in \mathbb{R}^{n \times n}\) og \(V \in \mathbb{R}^{d \times d}\) er ortogonale, og \(\Sigma\) er en diagonal matrix med ikke-negative tal i diagonalen, kaldet singulærværdier \(\sigma_1 \ge \sigma_2 \ge \cdots\).
Et vigtigt link til PCA er:
Dermed er de højre singulærvektorer (søjlerne i \(V\)) egenvektorerne for \(X^T X\) (og for kovariansmatricen op til skalering), dvs. de angiver hovedkomponenternes retninger. Desuden ser vi at singulærværdierne \(\sigma_i\) er ikke-negative tal, som opfylder \(\sigma_i = \sqrt{\lambda_i}\), hvor \(\lambda_i\) er den \(i\)’te største egenværdi af den symmetriske matrix \(X^T X\) (bemærk at \(X^T X\) faktisk er PSD, så egenværdierne er ikke-negative).
I den næste opgave skal vi beregne den første hovedretning ved hjælp af SVD og sammenligne den med den retning, man finder via variansmaksimerings-perspektivet.
U, S, Vt = np.linalg.svd(X_big, full_matrices=False)
w_svd = Vt[0] # Først højre singulære vektor (hovedretning)
print("Første hovedretning fra SVD:", w_svd)
print("Varians i denne retning:", variance_in_direction(X_big, w_svd))
Første hovedretning fra SVD: [-0.85193742 -0.52364361]
Varians i denne retning: 6.981964740988465
Opgave 2.3b#
Sammenlign denne retning med den, der gav dig den største varians ovenfor. Stemmer de overens (op til et fortegn)?
Hvis vi approksimerer hvert punkt \(( \pmb{x}_i )\) ved at projicere det ind på \(( \pmb{w} )\):
så bliver den samlede rekonstruktionsfejl:
Altså, minimering af rekonstruktionsfejlen er ækvivalent med maksimering af den projicerede varians, da \(\|X\|_F^2\) ikke afhænger af \(\pmb{w}\), dvs. udtrykket reduceres til:
Så de to metoder til at finde hovedkomponenter er faktisk ækvivalente!
Opgave 2.4*#
Implementér en lille funktion, der beregner \(E(\pmb w)\), og verificér, at en mindre fejl svarer til en højere varians:
def reconstruction_error(X, w):
# TO DO
pass
w_rand = np.random.randn(2)
print("Fejl (random dir):", reconstruction_error(X_big, w_rand))
print("Fejl (PCA dir): ", reconstruction_error(X_big, w_svd))
Fejl (random dir): None
Fejl (PCA dir): None
Numerisk verificering#
Opgave 2.5#
Brug følgende kode til at verificere, at optimiseringsformuleringen giver samme resultat som standard PCA.
import numpy as np
# Generér centreret tilfældig data
np.random.seed(42)
X_big = np.random.randn(100, 3)
C = X_big.T @ X_big / (X_big.shape[0] - 1)
# Analytisk PCA via egendekomposition
eigvals, eigvecs = np.linalg.eigh(C)
eigvecs = eigvecs[:, np.argsort(eigvals)[::-1]]
# Definér variansfunktionen
def variance(u):
u = u / np.linalg.norm(u)
return u.T @ C @ u
# Gittersøgningseksempel (blot for intuitionen skyld)
angles = np.linspace(0, 2*np.pi, 200)
variances = [variance(np.array([np.cos(a), np.sin(a), 0])) for a in angles]
# Sammenlign retningen, der giver maksimal varians, med top-egenvektoren
print("Første egenvektor:", eigvecs[:, 0])
Første egenvektor: [-0.1148552 -0.39297648 0.91234739]
Opgave 2.6#
Forklar, hvorfor PCA kan forstås som en sekvens af optimeringsproblemer.
Beskriv rollen af egenværdier og egenvektorer i denne formulering.
Diskutér, hvordan denne formulering kan udvides til komplekse data.
Afsnit 3: Hovedkoordinatanalyse (Principal Coordinate Analysis)#
Som et alternativ til PCA vil vi betragte Principal Coordinate Analysis (PCoA), også kaldet Multidimensional Scaling (MDS), som kan bruges til at reducere dimensionen af data baseret på dissimilariteter. Metoden er især nyttig inden for økologi, genetik og samfundsvidenskab, hvor forskellen mellem datapunkter har større tilgængelighed og/eller betydning end direkte målinger.
Igen betragter vi en vilkårlig reel datamatrix \(X \in \mathbb{R}^{n \times d}\), men denne gang sigter vi mod at konstruere en dissimilaritetsmatrix \(D \in \mathbb{R}^{n \times n}\), hvor \((i,j)\)-elementet \(d_{ij}\) svarer til dissimilariteten mellem det \(i\)-te og \(j\)-te datapunkt. Da vi vil arbejde med biologiske data, vælger vi Bray-Curtis (BC)-afstanden som dissimilaritetsmål, defineret ved
hvor \(x_{ik}\) betegner det \(k\)-te element af det \(i\)-te datapunkt. Hvis \(\pmb{x}_i = \pmb{x}_j = \pmb{0}\), definerer vi blot \(d_{ij}=0\). BC-afstanden anvendes typisk i økologi eller biologi ved sammenligning af \(n\) forskellige lokaliteter, som indeholder \(d\) forskellige arter. Hvis data kun indeholder ikke-negative værdier, ligger BC-afstanden mellem 0 og 1, hvor 0 betyder perfekt lighed (dvs. hver lokalitet indeholder samme antal af hver art), og 1 betyder maksimal dissimilaritet.
Opgave 3.1#
Vis, at hvis \(x_{ik} \geq 0\) for \(i = 1,\dots,n\) og \(k = 1,\dots,d,\) så er \(d_{ij} \in [0,1]\) for alle \(i,j\).
Opgave 3.2#
En alternativ måde at skrive BC-afstanden på er
Vis, at dette er ækvivalent med ovenstående definition.
Hint
Betragt de to tilfælde \(x_{ik} \geq x_{ij}\) og \(x_{ik} < x_{jk}\) for hvert led i summen.
Opgave 3.3*#
Ud fra definitionen er det klart, at \(D\) er symmetrisk, men det viser sig, at den kan have negative egenværdier. Vis, at hvis \(D \neq 0\), så har \(D\) mindst én negativ egenværdi.
Hint
Betragt vektoren \(\pmb{v}=\pmb{e}_i-\pmb{e}_j\), hvor \(\pmb{e}_k\) er en enhedsvektor med 1 i det \(k\)-te element og 0 ellers.
På samme måde som ved PCA skal vi centrere vores data, før vi beregner egenvektorer og egenværdier. Da vi nu betragter en symmetrisk matrix, vil vi have middelværdien til at være nul både i hver kolonne og hver række. Dette opnås ved såkaldt dobbeltcentrering, som giver matricen \(B\), defineret ved
hvor \(D^{(2)}\) er den elementvist kvadrerede afstandsmatrix \(D^{(2)}_{ij}=d_{ij}^2\), og \(H\) er \(n \times n\)-centreringsmatricen, defineret som
hvor \(\mathbf{1} \in \mathbb{R}^n\) er en vektor bestående af lutter ettaller.
Opgave 3.4#
Relatér dobbeltcentreringen til det, vi gjorde i normaliseringstrinnet før PCA. Bekræft, at hver række og hver kolonne i \(B\) har middelværdi nul.
Opgave 3.5#
Vis, at \(B\) tillader en spektral dekomposition
Hvad kan der konkluderes om \(B\)’s egenværdier?
Opgave 3.6#
Lav en Python-funktion, som tager en datamatrix \(X\) som input og returnerer centreringsmatricen \(B\). Test din funktion på et “overskueligt” datasæt ved at plotte egenrummene tilhørende de to egenvektorer med de største egenværdier.
En “overskueligt” datasæt kan være Iris-datasættet eller et endnu simplere datasæt i 2D som fx:
X = np.array([
[2.5, 2.4],
[0.5, 0.7],
[2.2, 2.9],
[1.9, 2.2],
[3.1, 3.0],
[2.3, 2.7],
[2.0, 1.6],
[1.0, 1.1],
[1.5, 1.6],
[1.1, 0.9],])
Med vores dobbeltcentrering på plads er vi nu klar til at beregne en lavdimensionel repræsentation af vores data. Vi vælger en dimension \(k\) og beregner
hvor \(\Lambda_k\) indeholder de \(k\) største egenværdier af \(B\), og \(Q_k\) de tilsvarende egenvektorer.
Opgave 3.7#
Lav en Python-funktion, som tager en datamatrix \(X\) og \(k\) som input og returnerer den lavdimensionelle PCoA-repræsentation \(Z\). Bemærk, at vi i dette tilfælde kan få negative egenværdier, så find en måde at håndtere dette på. Test din implementering på det lille datasæt, og plot resultatet. Sammenlign med PCA-plottet.
Opgave 3.8*#
Vis, at PCoA er ækvivalent med PCA (op til en skalering), hvis vi bruger den sædvanlige euklidiske afstand i stedet for Bray-Curtis-afstandsfunktionen. For enkelhedens skyld kan du antage, at \(X\) har søjlemiddelværdi nul.
Hint
Vi ønsker at vise, at
hvor \(E_B,E_C\) indeholder normerede egenvektorer for henholdsvis \(B=-\frac{1}{2}H D^{(2)}H\) og \(C = \frac{1}{n-1}X^TX\), og \(\Lambda_B\) indeholder egenværdierne for \(B\).
Hint
Vis, at \(B = XX^T\) ved brug af \(d_{ij}^2 = \|x_i-x_j\|_2^2\) og \(H\textbf{1} = \textbf{1}^TH = 0\).
Hint
Vis, at \(Xe_c\) er en egenvektor for \(B\) med egenværdien \((n-1)\lambda_c\) (du kan se bort fra faktoren \(n-1\)).
Afsnit 4: Arbejde med mikrobiomdata fra virkeligheden#
Med det grundlæggende på plads er det tid til at teste vores metoder på et virkeligt datasæt. Vi betragter de biologiske genus-data, som er gemt i den vedhæftede csv-fil. Rådataene indeholder lokationsoptællinger af forskellige bakterieslægter i 13 forskellige typer jordprøver: tre af typen uberørt græsland (P5, P8 og P9), tre af hvedetypen (S3, S7 og S9) samt majs (S1), rug (S4), byg (S5), raps (S6), græsfrø (S8) og lucerne (S10), med fem replikationer på hver lokalitet. Datasættet er højdimensionelt, og vores udviklede værktøjer (PCA og PCoA) vil blive anvendt til at lave lavdimensionelle visualiseringer. Disse kan i sidste ende bruges til at udtrække nyttig information om bioaktiviteten på artsniveau.
Opgave 4.1#
Download filen ALL_16S.csv. Indlæs dernæst csv-filen ved brug af
pandas-biblioteket, og konvertér den til en NumPy-array (en matrix) af passende størrelse. Hvad er antallet af datapunkter \(n\) og dimensionen \(d\) af hvert datapunkt? Hvad repræsenterer \(d\)?
Hint
For at læse csv-filen med pandas kan det være nødvendigt at ændre encoding of afgrænser (delimiter).
Opgave 4.2#
På baggrund af artiklen: Identification and Differentiation of Pseudomonas Species in Field Samples Using an rpoD Amplicon Sequencing Methodology og andre kilder, du finder online, skal du give en kort beskrivelse af relevansen af at profilere bakterier som dem i det givne datasæt. Beskriv mere specifikt nyttigheden af forskellige slægter såsom Pseudomonas til bioteknologiske formål, og omtal, hvorfor det kan være vigtigt at profilere dem på forskellige taksonomiske niveauer (fx slægts- eller artsniveau sammenlignet med familieniveau). Nævn også, hvordan PCA/PCoA kan hjælpe med denne profilering.
Opgave 4.3#
Diskuér, hvorfor Bray-Curtis-afstandsfunktionen er passende at anvende på dette datasæt. Er det nødvendigt at normalisere, før der køres PCoA?
Opgave 4.4#
Udfør PCA og PCoA med \(k=1,2,3\), og visualisér resultaterne på en passende måde med forskellige farver og labels for de forskellige lokaliteter. Beskriv de forskelle, du ser mellem de to metoder, og sammenlign med følgende figur* fra artiklen.
Bemærk, at forfatterne i artiklen anvendte såkaldt non-metric multidimensional scaling (NMDS), som er en generalisering af PCoA, der er velegnet til at håndtere komplekse datasæt og afdække relative relationer i ikke-lineære data. Selvom metoden er anderledes, bør du kunne producere et nogenlunde lignende plot.
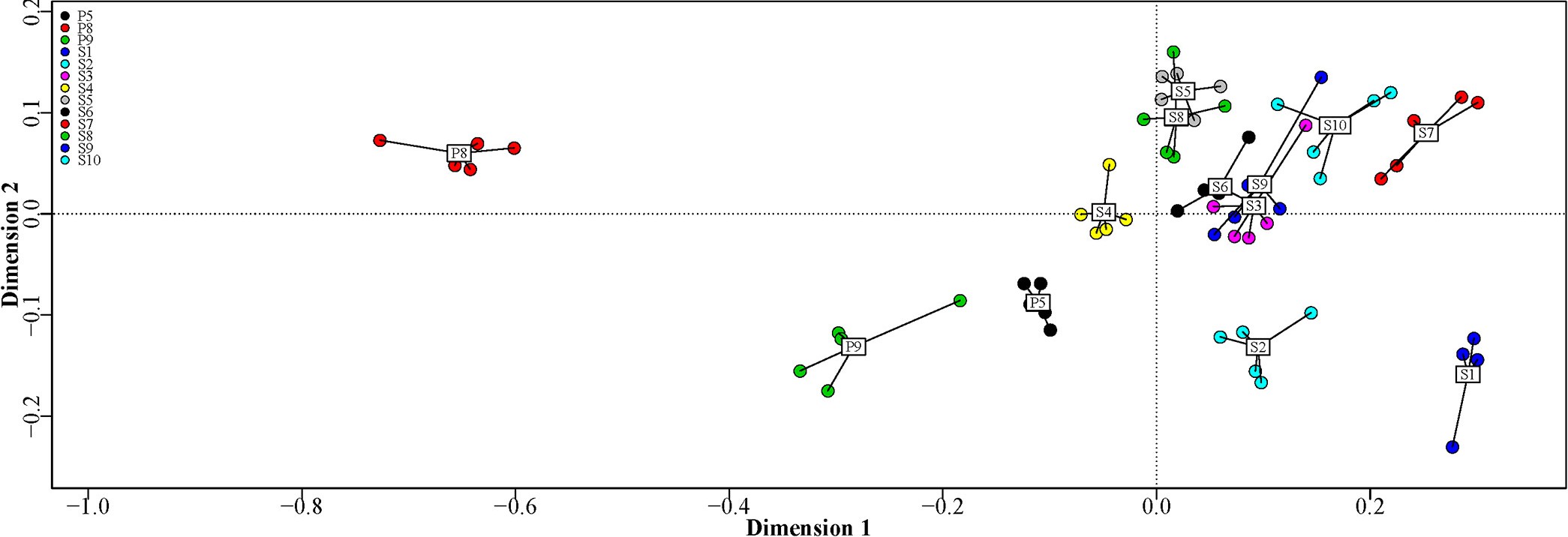
Opgave 4.5#
Plot egenværdierne for PCA- og PCoA-resultaterne på passende vis. Hvor høj en værdi af \(k\) giver det mening at bruge?
Opgave 4.6#
Diskutér mulige biologiske forklaringer på ovenstående resultater. Din diskussion bør nævne årsager til isolerede klynger i den lavdimensionelle repræsentation. Det kan være nyttigt at inddrage de øvrige figurer i artiklen for mere information om dataene. Husk at skelne mellem målemetoderne i artiklen - kun de figurer, hvor 16S undersøges, er relevante.